鋰金屬電池(LMBs)因其高能量密度被視為下一代儲能設備。然而,目前的電解質體系在溶劑化調控方面存在不足,導致了緩慢的鋰離子傳輸和不可控的鋰枝晶生長,限制了鋰金屬電池應用。通過降低Li+與溶劑的親和力而形成的弱溶劑化環境有利于改善LMBs電解質的性能。這是因為當陰離子進入Li+溶劑化鞘內時,將會形成由陰離子衍生的富含無機組分的固態電解質界面(SEI)。然而,弱溶劑化調控難免對鋰鹽解離產生影響,導致電解質的離子電導率降低,從而影響電解質的離子傳輸性能。因此,如何合理調節溶劑化以實現鋰離子的快速傳輸和穩定的固態電解質界面(SEI)之間的平衡對優化電池穩定性能至關重要。
【工作簡介】
近日,武漢理工大學木士春、曾煒豪團隊及廖小彬博士等研究者通過原位聚合策略設計并構建了三種具有高、中、低Li+-溶劑結合強度的凝膠聚合物電解質(GPEs)。其中,具有中等Li+-溶劑結合強度的凝膠聚合物電解質(MB-GPE)促進了陰離子衍生的溶劑化結構,形成了富含無機成分(如LiF)的SEI,有效抑制了界面副反應,并加速了界面反應動力學。此外,MB-GPE對鋰鹽解離的影響較小,具有優異的離子電導率。相比之下,具有高和低Li+-溶劑結合強度的GPEs在LMBs中表現出較差的循環性能。這主要是由于SEI穩定性和Li+傳輸受到顯著限制。研究證實,設計具有中等Li+-溶劑結合強度的凝膠聚合物電解質是一種有效策略,能夠實現快速的Li+傳輸并促進穩定SEI層的形成,從而確保鋰金屬電池的高比容量和長期穩定性。該文章發表在國際頂級期刊Energy & Environmental Science上。張少杰和李忠澎為本文共同第一作者。
【內容表述】
在這項工作中,為了探尋溶劑化調控與電解質性能之間的關系,作者采用原位聚合策略設計、構建了具有不同Li+-溶劑結合力的GPEs。他們合成了三組具有不同氟化組分的凝膠電解質,即碳酸乙烯酯(EC)和碳酸甲乙酯(EMC)、氟化碳酸乙烯酯(FEC)和2,2,2-三氟乙基碳酸甲酯(FEMC)及二氟碳酸乙烯酯(DFEC)和FEMC,分別具有高Li+-溶劑結合力(HB-GPE)、中等Li+-溶劑結合力(MB-GPE)和低Li+-溶劑結合力(LB-GPE)。在本項工作中,與鋰金屬具有良好相容性的聚乙二醇二丙烯酸酯(PEGDA)被選擇作為聚合單體。
3.1 凝膠聚合物電解質的設計
在具有高Li+-溶劑結合力的凝膠電解質(HB-GPE)中,由于溶劑較強的親和力,鋰鹽可實現充分解離。但溶劑過多地進入Li+的溶劑化鞘中時,在遷移至金屬鋰表面時容易生成含大量有機組分的SEI,而且鋰枝晶生長亦較快,不利于鋰金屬的穩定循環。而在具有低Li+-溶劑結合力的凝膠電解質(LB-GPE)中,由于溶劑的溶劑化能力較差,Li+仍與陰離子緊密結合,大量鋰鹽未解離,導致離子電導率大幅降低。但由于較多的陰離子可遷移至Li+的溶劑化鞘中,界面性能有所改善。在具有適度Li+-溶劑結合力的凝膠電解質(MB-GPE)中,由于溶劑具有適度親和力,Li+既能擺脫陰離子的束縛,鋰鹽得到充分解離,賦予電解質高的離子電導率,陰離子也能順利遷移至Li+的溶劑化鞘中,從而衍生出由陰離子主導且富含無機組分(例如LiF)的SEI,可有效抑制鋰枝晶的生長。在實現金屬鋰的高穩定循環的同時,還可實現鋰離子的快速遷移。
本工作首先對三種具有不同Li+-溶劑結合的凝膠聚合物電解質進行了模擬計算。從EC到FEC再到DFEC,Li+與不同溶劑的結合力數值從-2.2降至-2.0 eV,再降至-1.81 eV。此外,從EMC到FEMC,結合力數值從-2.0降至-1.66 eV。計算結果表明,隨著氟化程度的增加,Li+-溶劑結合力逐漸降低。采用分子動力學(MD)模擬分析Li+-溶劑結合力與溶劑化結構之間的關系。在第一溶劑化鞘層(r=2 A)中,對于HB-GPE,Li+與EC的配位數為0.4,Li+與EMC的配位數為0.29;對于MB-GPE,Li+與FEC的配位數為0.19,Li+與FEMC的配位數為0.06;對于LB-GPE,Li+與DEFC的配位數為0.08,Li+與FEMC的配位數為0.1。因此,隨著凝膠電解質中Li+與溶劑結合力的減少,在第一溶劑化鞘中,Li+/溶劑的總配位數逐漸減少,而Li+/TFSI-的配位數逐漸增加。徑向分布函數(RDFs)結果進一步證實,隨著溶劑分子氟化程度的增加,在第一溶劑化鞘層(r=2 A)中,Li+與溶劑分子的相互作用逐漸減弱(EC>FEC>DFEC,EMC>FEMC),更多的陰離子進入到Li+的溶劑化結構中,有利于形成由陰離子衍生的富含無機物的SEI。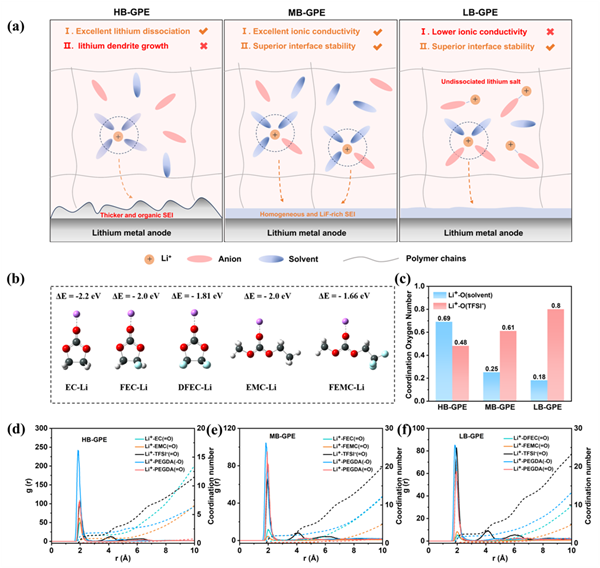
圖1 凝膠聚合物電解質設計和溶劑化結構研究。(a) HB-GPE、MB-GPE和LB-GPE的溶劑化結構和SEI演變過程。(b) 不同溶劑與Li+的結合力計算。(c) 不同凝膠電解質中Li+與溶劑和TFSI-的氧原子配位數變化。(d,e,f)由MD模擬得到的HB-GPE、MB-GPE和LB-GPE的徑向分布函數。
3.2 物化性能評估
采用密度泛函理論(DFT)對三種凝膠聚合物電解質所使用的溶劑和鋰鹽的最高占據分子軌道(HOMO)能級和最低未占據分子軌道(LUMO)能級計算。從EC到FEC,再到DFEC,HOMO和LUMO能值逐漸降低;從EMC到FEMC,HOMO和LUMO能值也逐漸降低。LiTFSI的LUMO能級最低(-1.43 eV),更有利于在鋰陽極處被還原分解,形成富含LiF無機組分的SEI。
通過Raman拉曼光譜和液態核磁NMR測試分析Li+在HB-GPE、MB-GPE和LB-GPE的溶劑化結構中的解離狀態。LiTFSI的光譜可擬合成三個部分:自由TFSI-(位于738.9 cm-1,未配位的TFSI-)、接觸離子對(CIP,742.7 cm-1,與一個Li+結合的TFSI-)和聚集體(AGG,747.9 cm-1,與兩個或更多Li+結合的TFSI-)。LB-GPE中存在少量游離TFSI-信號,表明LB-GPE中存在大量未解離的Li+。MB-GPE中存在較多的游離TFSI-信號和CIP信號,表明鋰鹽已充分解離,有利于形成由陰離子主導的溶劑化結構。進一步采用7Li NMR分析溶劑化結構中Li+周圍的化學環境。與HB-GPE相比,MB-GPE和LB-GPE的7Li峰向左移,表明Li+周圍的電荷屏蔽程度降低,有利于鋰離子的快速遷移。這證明具有適度Li+-溶劑結合力的MB-GPE可同時實現LiTFSI的完全解離和Li+的快速遷移。雖然HB-GPE具有最佳的鋰解離狀態,但鋰離子遷移受阻;而低Li+-溶劑結合力的LB-GPE則會大幅阻礙LiTFSI的解離。
測試結果表明,MB-GPE在室溫下具有高的離子電導率(1.95×10-3 S cm-1),居于LB-GPE(6.73×10-4 S cm-1)和HB-GPE(3.84×10-3 S cm-1)之間。MB-GPE的遷移活化能(0.151 eV)略高于LB-GPE的0.143 eV,而低于HB-GPE(0.193 eV)。鋰離子遷移勢壘的降低有利于Li+快速遷移。MB-GPE和LB-GPE的氧化電位分別為5.12 V和5.23 V,高于HB-GPE的4.08 V,表明MB-GPE擁有高的抗氧化性能。在4.2至4.7 V的電壓范圍內,使用MB-GPE和LB-GPE的Li||NCM811電池均出現穩定的漏電流,表現出優異的高壓穩定性。而使用HB-GPE的電池在恒壓充電至4.3 V時電流顯著增加,表明在4.3 V高壓下HB-GPE的穩定性較差。此外,MB-GPE的Li+遷移數為0.62,分別高于HB-GPE(0.58)和LB-GPE(0.31)。較高的Li+遷移數表明MB-GPE的界面極化較低,降低了濃差極化的影響,從而促進鋰的均勻沉積。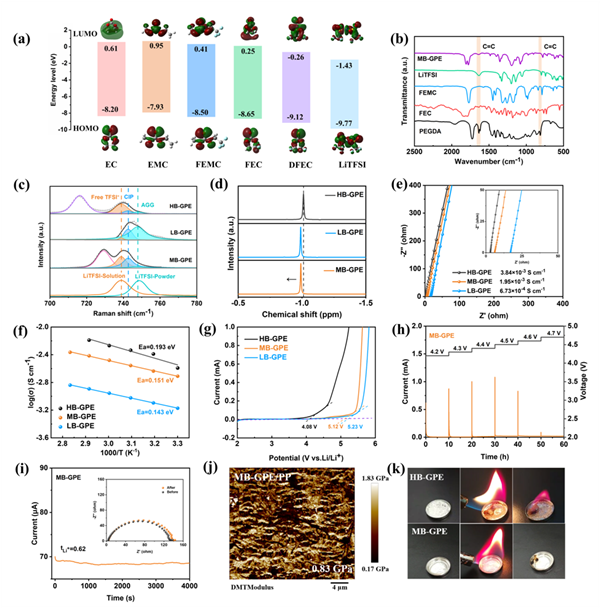
圖2 HB-GPE、MB-GPE和LB-GPE物化特性。(a) EC、EMC、FEMC、FEC、DFEC和LiTFSI的HOMO和LUMO能級。(b) MB-GPE、LiTFSI、FEMC、FEC和PEGDA傅立葉變換紅外光譜,波長范圍為500-2500 cm-1。(c) HB-GPE、MB-GPE和LB-GPE的拉曼光譜。(d) HB-GPE、MB-GPE和LB-GPE的7Li NMR光譜。(e) HB-GPE、MB-GPE和LB-GPE離子電導率。(f) HB-GPE、MB-GPE和LB-GPE的阿倫尼烏斯擬合。(g) HB-GPE、MB-GPE和LB-GPE的LSV曲線。(h) MB-GPE電化學浮動分析。(i) 使用MB-GPE的鋰對稱電池的極化曲線。插圖顯示極化前后的奈奎斯特圖。(j) MB-GPE/PP復合隔膜的楊氏模量圖。(k) HB-GPE和MB-GPE可燃性測試。
3.3 金屬鋰界面穩定性分析
使用HB-GPE、LB-GPE和MB-GPE等三種凝膠聚合物電解質組裝了Li||Li對稱電池,對金屬鋰在鋰沉積和剝離過程中的界面穩定性進行評估。測試電流密度為0.5 mA cm-2,容量為0.5 mAh cm-2。Li|MB-GPE|Li對稱電池穩定循環超過3200小時后仍保持約30 mV的低過電位,具有優異的循環穩定性能。相比之下,使用HB-GPE和LB-GPE的Li||Li對稱電池在循環900小時和1367小時后分別出現短路,極化電壓驟減,表明此時金屬鋰發生嚴重副反應,界面穩定性較差。進一步測試臨界電流密度(CCD)以評估HB-GPE、LB-GPE和MB-GPE抑制鋰枝晶的能力。MB-GPE的CCD為3.5 mA/cm2,而HB-GPE和LB-GPE的CCD分別為1.5和2 mA/cm2。這表明MB-GPE可承受更大的電流密度,在抑制鋰枝晶生長方面的能力最強。
分別使用HB-GPE、LB-GPE和MB-GPE組裝了Li||Cu電池,進一步考察其鋰沉積和剝離過程中的庫侖效率(CE)。使用MB-GPE和LB-GPE的鋰銅電池的平均CE值分別為93.42%和93.64%,均高于HB-GPE(65.97%)。使用MB-GPE的Li||Cu電池在循環第1、50和150圈時的極化電壓分別為82、40和46 mV。較小的極化電壓表明MB-GPE在鋰沉積-剝離過程中具有出色的穩定性。而使用HB-GPE的Li||Cu電池在循環第1和50圈時的極化電壓分別為108和99 mV,隨后庫倫效率迅速降低。使用LB-GPE的Li||Cu電池在循環第1、50和150圈時的極化電壓分別為134、97和161 mV。其較大的極化電壓與其較差的鋰沉積形態相對應。進一步分析銅箔表面沉積的鋰的形態和厚度可知,在使用HB-GPE的銅箔上可觀察到疏松多孔的針狀鋰枝晶,表明HB-GPE與鋰金屬發生了嚴重的副反應,并產生大量死鋰;當使用MB-GPE時,銅箔上沉積的鋰表面平整致密,沒有可見的枝晶;當使用LB-GPE時,沉積在銅箔上的鋰呈現出帶有凹坑的塊狀形態,表明存在不均勻的鋰沉積。截面SEM觀察結果表明,使用MB-GPE和LB-GPE后的沉積鋰的厚度分別為16.8和16.9 μm,小于HB-GPE的48.6 μm。這表明MB-GPE能夠促進鋰離子通量的均勻分布并調節鋰成核,最終實現鋰的均勻沉積。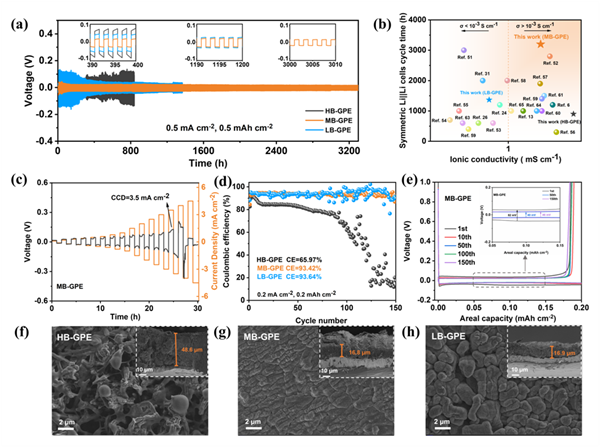
圖3 鋰沉積/剝離性能評估和鋰沉積形態表征。(a)基于HB-GPE、MB-GPE和LB-GPE的Li||Li對稱電池在0.5 mA cm-2和0.5 mAh cm-2條件下的循環性能。(b)本研究中的GPE與最近報道的其他GPE在離子電導率和對鋰穩定性方面的比較。(c)Li|MB-GPE|Li對稱電池的臨界電流密度測試。(d)電流密度為0.2 mA cm-2、容量為0.2 mAh cm-2時鋰在銅箔上的沉積/剝離庫侖效率。(e)基于MB-GPE的Li||Cu電池的電壓-容量曲線,插圖顯示了第1、50和150圈時的電壓-容量曲線。在基于(f)HB-GPE、(g)MB-GPE和(h)LB-GPE電池中,在1 mA cm–2和5 mAh cm–2條件下鋰在銅箔表面的沉積形貌;插圖是使用HB-GPE、MB-GPE和LB-GPE后鋰沉積的橫截面形態。
3.4 鋰金屬電池的循環性能分析
以磷酸鐵鋰(LiFePO4)為正極,鋰金屬為負極,組裝成Li||LiFePO4鋰金屬電池,并測試電池在不同倍率下的循環性能。Li|MB-GPE|LiFePO4電池在0.5 C下的初始放電比容量為144.6 mAh g-1;在循環780圈后,其放電比容量為122 mAh g-1,平均CE值為99.99%,容量保持率為84.4%。相比之下,Li|HB-GPE|LiFePO4和Li|LB-GPE|LiFePO4的初始放電比容量分別為126.7和139.7 mAh g-1,循環500圈后的容量保持率分別為46.8%和72.3%。Li|MB-GPE|LiFePO4電池在更大倍率(1 C)下仍具有優異的循環性能。
為了評估MB-GPE與高鎳正極材料(NCM811)的兼容性,組裝了Li||NCM811鋰金屬電池,并在3.0-4.3 V的電壓范圍內測試其循環性能。使用MB-GPE的Li||NCM811電池在0.5 C下的初始放電比容量為157 mAh g-1。在循環400圈后,其放電比容量降至125.8 mAh g-1,但容量保持率仍高達80.1%。而Li|LB-GPE|NCM811電池的初始放電比容量為135.8 mAh g-1,循環230次后容量保持率降為52.1%。值得注意的是,僅僅循環10次后,Li|HB-GPE|NCM811電池的放電比容量就從145.2下降到77.5 mAh g-1,難以在高電壓下正常工作。這個測試結果也與HB-GPE較低的電化學窗口和較差的電化學浮動測試結果相對應。為了進一步證明MB-GPE在實際應用中的潛力,進一步將其與高負載的NCM811正極片(負載量:9.2 mg cm-2)和超薄的鋰箔負極(厚度:50 μm)組裝成軟包電池。軟包電池在0.2 C倍率下可穩定循環20個周期,容量保持率為84%,并表現出優越的安全性能。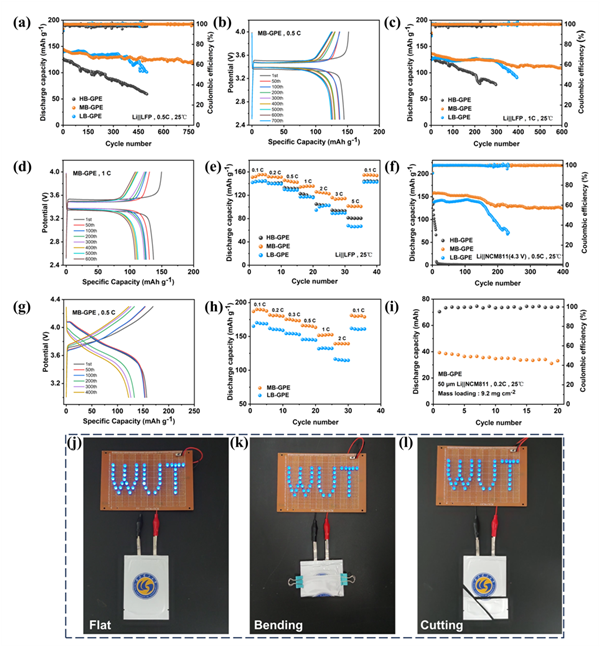
圖4 基于HB-GPE、MB-GPE和LB-GPE電池的鋰金屬電池循環性能。(a) 基于三種凝膠聚合物電解質的Li||LFP電池在0.5 C下的循環性能。(b)基于MB-GPE的Li||LFP電池在0.5 C倍率,4 V截止電壓下的放電/充電電壓曲線。(c) 基于三種凝膠聚合物電解質的Li||LFP電池在1 C下的循環性能。(d) 基于MB-GPE的Li||LFP電池在1 C倍率,4 V截止電壓下的放電/充電電壓曲線。(e)基于三種凝膠聚合物電解質的Li||LFP電池的倍率性能,以及(f)Li||NCM811電池在0.5 C下的循環性能。(g)基于MB-GPE的Li||NCM811電池在0.5 C倍率,4.3 V截止電壓下的放電/充電電壓曲線。(h)基于三種凝膠聚合物電解質的Li||NCM811電池的倍率性能。(i)基于MB-GPE的Li||NCM811軟包電池的循環性能。基于MB-GPE的Li||NCM811軟包電池在不同狀態的安全測試:(j)鋪展;(k)彎折;(l)剪切。
3.5 電極界面分析
基于三種凝膠聚合物電解質的Li||LiFePO4電池在0.5 C下循環20次后,進一步采用X射線光電子能譜(XPS)對其鋰金屬表面進行深度蝕刻,以分析SEI組分。在C 1s光譜中,基于HB-GPE和LB-GPE電池的金屬鋰表面含有大量的C-O和C=O有機成分,且隨著刻蝕深度的增加,C-O和C=O有機成分仍大量存在。這表明基于HB-GPE和LB-GPE電池的金屬鋰發生了嚴重的副反應,從而產生較多有機組分。相反,基于MB-GPE電池的金屬鋰中,可以觀察到少量的C-O和C=O成分,且隨刻蝕深度增加,含量逐漸減少;此外,金屬鋰中的Li2CO3無機成分較多,含量分布均勻。在O1s光譜中,與HB-GPE和LB-GPE相比,隨著蝕刻深度的增加,基于MB-GPE電池的SEI層中觀察到了Li2O無機組分,但沒有發現RO-Li有機成分。在F1s光譜中,與HB-GPE和LB-GPE相比,基于MB-GPE電池的金屬鋰表面僅含有LiF無機氟化物,表明形成了富含Li2CO3、Li2O、LiF等無機組分的SEI層。多種無機組分的協同作用,提高了電池的電化學穩定性,抑制了鋰枝晶生長,并構筑了均勻的Li+擴散通量,賦予了鋰金屬負極高的循環壽命。
基于MB-GPE電池,進一步采用了飛行時間二次離子質譜法(TOF-SIMS)探究其SEI中無機組分的三維含量分布。隨著濺射時間的增加,基于HB-GPE電池的金屬鋰中C2HO-有機組分的含量始終多于LiF2-,無機組分占少量。與HB-GPE相比,基于MB-GPE電池的薄SEI層中同時富含LiF2-(來自LiF)、Li2CO3-和LiO2-離子;而且C2HO-有機組分的含量低于LiF2-,無機組分占主導。這些研究結果表明,MB-GPE促進了富含Li2CO3、Li2O和LiF無機成分的SEI層的形成,有利于抑制鋰枝晶的生長,從而實現鋰金屬電池的長期穩定循環。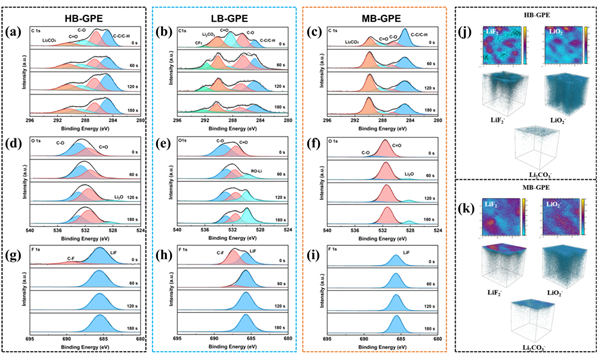
圖5 SEI層的界面化學分析:基于(a)HB-GPE、(b)LB-GPE和(c)MB-GPE電池的鋰金屬陽極循環20次后C 1s的XPS深度分析;基于(d)HB-GPE、(e)LB-GPE和(f)MB-GPE電池的鋰金屬陽極循環20次后O 1s的XPS深度分析;基于(g)HB-GPE、(h)LB-GPE和(i)MB-GPE電池的鋰金屬陽極循環20次后F 1s的XPS深度分析。基于(j)HB-GPE和(k)MB-GPE電池的鋰金屬表面的代表性二次離子的TOF-SIMS二維和三維映射圖像。
【核心結論】
本項工作中,研究員人員設計、合成了具有適度Li+-溶劑結合力的含氟凝膠聚合物電解質(MB-GPE),并系統評估了具有高、適中、低Li+-溶劑結合力凝膠聚合物電解質的電化學性能。結果表明,引入氟化溶劑FEC和FEMC的MB-GPE能有效地平衡弱溶劑化調控和鋰離子傳輸之間的矛盾;此外,該電解質還有助于形成富含LiF等無機組分的SEI,極大改善了鋰金屬負極的循環性能。MB-GPE具有1.95×10-3 S cm-1的高離子電導率、0.62的鋰離子遷移數和5.12 V的寬電化學窗口。基于MB-GPE的Li||Li對稱電池在0.5 mA cm-2的電流密度下實現了超過3200小時的穩定循環,而Li|MB-GPE|Cu電池在0.2 mA cm-2的電流密度下展現出優異的鋰沉積/剝離性能和高的庫倫效率(93.42%)。當MB-GPE應用于LiFePO4和NCM811多種正極的鋰金屬電池時,其循環可逆性顯著提高。此外,使用MB-GPE組裝的Li||NCM811軟包電池具有良好的循環性能和優異的安全性能。本工作所提出的溶劑化結構策略為設計高性能凝膠聚合物電解質及高性能固態電池的開發提供了一個新的思路。
【文獻詳情】
Shaojie Zhang#, Zhongpeng Li#, Yixin Zhang, Xuanpeng Wang, Peiyang Dong, Saihai Lei, Weihao Zeng*, Juan Wang, Xiaobin Liao*, Xingye Chen, Dongqi Li, Shichun Mu*. Moderate Li+-Solvent Binding for Gel Polymer Electrolytes with Stable Cycling toward Lithium Metal Batteries. Energy & Environmental Science, 2025, Doi:10.1039/D4EE05866F.