第一作者:蔣林濤、陳興寶
通訊作者:李蓓副教授、木士春教授
通訊單位:武漢理工大學
論文DOI:10.1021/acscatal.4c07602
全文速覽
近日,武漢理工大學木士春教授和李蓓副教授課題組合作設計了一種活化羥基誘導界面水的質子交換策略,用于高效堿性析氫反應(HER)。該研究通過電化學活化方法合成了一種具有核殼結構的R-Ru-Ni(OH)2催化劑,并成功在Ru表面引入了大量活化羥基(OHad)。研究表明,OHad的引入優化了Ru活性位點的電子/能帶結構和物理化學狀態,并顯著參與質子交換過程,改善了界面水分子的吸附和解離步驟,從而加速了催化動力學過程。具體而言,表面活化的OHad位點通過直接的OH-H2O質子交換優化了水的吸附路徑,并以低至0.035 eV的吉布斯自由能實現了吸附水物種的分解;而且,額外的OHad位點改善了*H的脫附行為,打破了OH阻礙的動力學限制,并雙向降低了決速步驟的能壘。在電流密度為10和100 mA cm-2堿性條件下,其HER過電位分別僅為26和81 mV,并具有出色的穩定性(在20 mA cm-2下達到100 h)。此外,作者進一步證明了金屬表面活化羥基-界面水質子交換策略的普適性。本研究從實驗和理論上證明了活化羥基對堿性析氫反應中界面水質子交換過程的調節,研究成果將為高效催化劑的合理設計提供指導。
背景介紹
催化劑界面關鍵中間體的化學狀態與微觀結構顯著影響電化學性能。對于堿性析氫反應(HER),存在著界面水的吸附與解離以及*H、*OH中間體的形成與脫附等復雜過程,嚴重阻礙了HER反應的進行。一般而言,堿性HER動力學過程比酸性低2-3個數量級。因此,改善堿性HER催化劑的中間體相互作用有利于調節活性位點本征活性,降低反應能壘,加快催化動力學。通常認為,在堿性HER反應路徑中,*OH/OH-的形成與脫附是不可避免的。如果采用通常改善其中間體行為的策略,催化過程中將會受到因*H/*OH競爭性吸附導致的動力學阻礙,不利于進一步優化堿性HER活性。相較而言,通過利用催化劑表面吸附的羥基物種來直接參與HER過程,有望進一步改善催化反應路徑,提高本征動力學。這是因為,*OH是一種電子有利的質子受體,相較于純的原子級金屬活性中心更有利于與界面水物種相互作用,從而促進水吸附過程。另一方面,由于*OH直接參與反應形成額外的OH位點,避免了OH脫附步驟,因而加速了隨后的水解離與*H脫附過程。因此,*OH位點直接參與HER反應可以雙向改善堿性析氫過程,有利于打破*H與*OH競爭性動力學障礙,從而極大提高本征活性。
本文亮點
1. 本文首先合成了一種核殼催化劑Ru-Ni(OH)2,然后通過原位電化學活化方式進一步合成了具有核殼結構的R-Ru-Ni(OH)2催化劑,同時實現了Ru表面活化OHad物種的引入,有效提升其堿性HER活性。系列測試亦證明了該策略的普適性。
2. 本研究從實驗和理論上證明了金屬表面的活化OHad與界面水發生了顯著的質子交換,實現了HER電子轉移路徑的優化,為高性能堿性HER催化機制設計提供了一種新穎的思路。
圖文解析

圖1 (a) R-Ru-Ni(OH)2制備的示意圖。(b, c) NF上Ru-Ni(OH)2和R-Ru-Ni(OH)2的SEM圖像。(d, e) Ru-Ni(OH)2和R-Ru-Ni(OH)2的HRTEM圖像。(f, g) 通過EDS映射得到的Ru-Ni(OH)2和R-Ru-Ni(OH)2相應的元素分布情況。
通過水熱合成及電化學CV活化重構等過程,獲得核殼催化劑R-Ru-Ni(OH)2/NF(圖1a)。通過SEM和TEM證明了通過水熱法成功合成了Ru-Ni(OH)2核殼納米結構材料(圖1b,c)。其中,Ru核的直徑約為80-100 nm,Ni(OH)2殼層約20 nm厚度。經電化學活化獲得的R-Ru-Ni(OH)2催化劑的結構沒有出現明顯的變化(圖1d,e)。其晶格條紋主要對應于晶體Ru,Ni(OH)2則更分布在外殼層。EDS表明Ru元素主要分布在Ru-Ni(OH)2納米球內部,而Ni、O分布在Ru-Ni(OH)2納米球表面。這些進一步證明了Ru-Ni(OH)2核殼結構的形成(圖1f, g)。
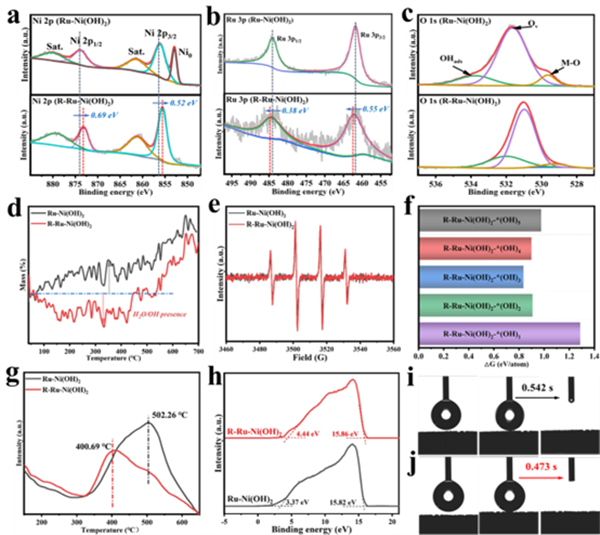
圖2 (a, b, c) R-Ru-Ni(OH)2和Ru-Ni(OH)2的Ni 2p、Ru 3p和O 2p的高分辨率XPS。(d) R-Ru-Ni(OH)2和Ru-Ni(OH)2在氬氣氛圍下的TG曲線。(e) R-Ru-Ni(OH)2和Ru-Ni(OH)2的EPR譜圖。(f) 不同數量OH吸附基團的R-Ru-Ni(OH)2-*(OH)x的形成能。(g) R-Ru-Ni(OH)2和Ru-Ni(OH)2的H2-TPD曲線。(h) R-Ru-Ni(OH)2和Ru-Ni(OH)2的UPS譜圖。(i) Ru-Ni(OH)2和 (j) R-Ru-Ni(OH)2的水接觸角測試。
XPS證明了催化劑經電化學活化后電子從Ru核轉移到Ni(OH)2殼層上,有利于加強Ru位點對羥基基團的吸附作用(圖2a,b)。同時,O 1s光譜亦證明催化劑活化后存在著更多的表面含O基團(圖2c)。此外,熱重曲線的下降(質量減少)驗證了催化劑中存在更多的結合水與羥基物種(圖2d)。從圖2e可以看出,活化后的催化劑表現出更強的EPR信號,表明R-Ru-Ni(OH)2形成了更多的羥基。接著,通過DFT計算解析了Ru-Ni(OH)2中Ru催化劑表面的羥基吸附動力學,表明OHad易吸附在Ru催化劑表面(圖2f)。H2-TPD證明了R-Ru-Ni(OH)2更利于H2脫附(圖2g),而且由UPS分析可知,R-Ru-Ni(OH)2還有利于中間體脫附(圖2h)。從圖2i, j的水接觸角分析可知,R-Ru-Ni(OH)2具有更優異的吸水性與親水能力。這些研究結果均與其表面存在的親水羥基基團有關。
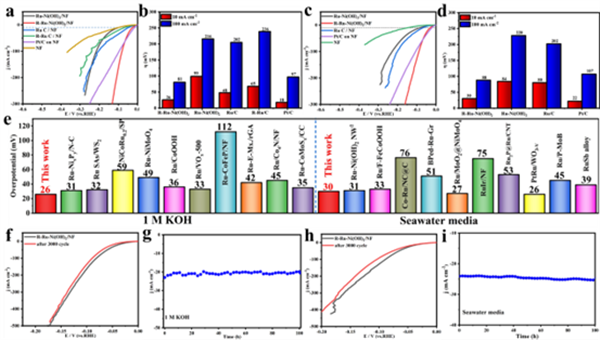
圖3 催化劑在 (a, b) 1 M KOH溶液和 (c, d) 海水介質中的LSV曲線及相應過電位(@10和100 mA cm-2)。(e) 在堿性介質中,R-Ru-Ni(OH)2與近期報道的釕基電催化劑在10 mA cm-2時析氫反應(HER)性能的比較。R-Ru-Ni(OH)2在 (f, g) 1 M KOH溶液和 (h, i) 海水介質中的3000次循環伏安穩定性和電流密度隨時間變化的曲線。
基于此,進一步研究了所制備催化材料在堿性條件下的析氫性能。由圖3a可以看出,在所有催化劑中,R-Ru-Ni(OH)2催化劑表現出最佳的活性:在電流密度分別為10與100 mA cm-2下過電位分別為26與81 mV,可與商業Pt/C性能(18,97 mV)相媲美(圖3a-e)。此外,R-Ru-Ni(OH)2亦具有優異的CV循環(3000圈)和i-t長期(100 h)穩定性(圖3f-i)。此外,羥基-界面水質子交換策略具有廣泛的通用性。對于Ru-M(OH)x/MF(M = Fe、NiFe、Cu)和M-Ni(OH)2/NF(M = Ir、Pt),電化學活化后其堿性HER性能普遍得到改善。![]()
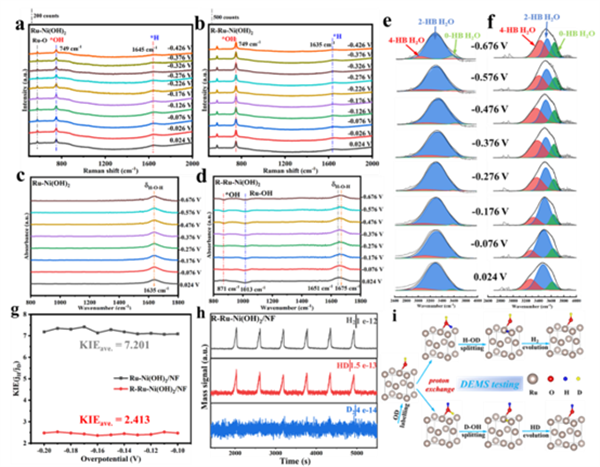
圖4 (a) Ru-Ni(OH)2和(b) R-Ru-Ni(OH)2在1 M KOH溶液中從0.024至-0.426 V vs.RHE的原位拉曼光譜。(c, e) Ru-Ni(OH)2和 (d, f) R-Ru-Ni(OH)2在從0.024至-0.676 V vs.RHE的原位衰減全反射表面增強紅外吸收光譜(ATR-FTIR)。(g) R-Ru-Ni(OH)2和Ru-Ni(OH)2的動力學同位素效應(KIE)值。(h) R-Ru-Ni(OH)2在1 M KOD/D2O溶液中經CV活化后,通過原位微分電化學質譜(DEMS)檢測的產氫信號。(i) 氘標記的R-Ru-Ni(OH)2的DEMS測試示意圖。
為了揭示R-Ru-Ni(OH)2堿性HER的反應機理與活化羥基的積極作用,采用原位光譜對活化前后Ru-Ni(OH)2的HER過程進行了研究。原位拉曼/紅外光譜(圖4a-f)證明,R-Ru-Ni(OH)2擁有更多的表面羥基參與反應,改善了OH中間體演變動力學,有利于界面水的質子交換過程(包括吸附與解離)。R-Ru-Ni(OH)2計算的KIE值更低(圖4g),表明HER中O-H更容易斷裂以及水解離的加速。DEMS測試結果分析表明,在含D的電解液標記之后,R-Ru-Ni(OH)2出現了明顯的HD信號。HD主要來源于KOD/D2O電解液中電化學活化標記的OD物種,通過質子交換與之后的HDO裂解-脫附,從而形成HD氣體。這些有力地證明了穩定的表面活化羥基能夠顯著參與OH-H2O質子交換反應,促進HER動力學(圖4h,i)。 ![]()
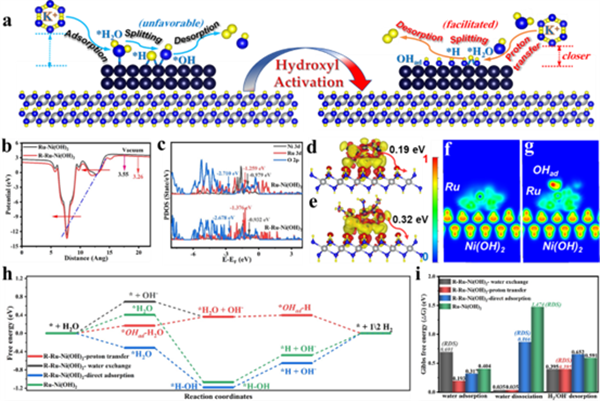
圖5 (a) Ru-Ni(OH)2在HER過程中活化前后電子轉移及反應中間體演變路徑的示意圖。(b) R-Ru-Ni(OH)2和Ru-Ni(OH)2的功函數。(c) R-Ru-Ni(OH)2和Ru-Ni(OH)2中Ru、Ni、O原子的部分態密度(PDOS)。(d) Ru-Ni(OH)2和 (e) R-Ru-Ni(OH)2的Bader電荷(DCD)。(f) Ru-Ni(OH)2和 (g) R-Ru-Ni(OH)2的電子局域函數(ELF)。 (h) R-Ru-Ni(OH)2以及Ru-Ni(OH)2中Ru位點在不同路徑下的HER反應的吉布斯自由能變化。 (i) R-Ru-Ni(OH)2和Ru-Ni(OH)2中Ru位點在不同路徑下的水吸附、水解離和氫/羥基解吸的能壘。
圖5a展示了Ru-Ni(OH)2核殼催化劑在羥基活化前后及HER過程中電子/中間體的轉移/演變路徑。以Ru-OHad為反應位點,可實現更低能壘的質子交換過程,從而促進液態水轉移到催化劑表面,同時在羥基位點存在的條件下加速水解離與*H脫附進程。通過WF、PDOS、DCD、ELF以及吉布斯自由能計算,證明了活化OHad位點的電子/能帶結構得到改善(圖5b-i)。表面活化的OHad位點直接參與并優化了OH-H2O質子交換反應,實現了水吸附并加速了后續水解離過程。同時,由于額外OHad位點的存在改善了*H脫附行為,顯著降低了決速步驟反應能壘,極大地提高了催化劑的反應活性。
總結與展望
總之,通過采用簡單的電化學活化策略成功將羥基物種引入到Ru-Ni(OH)2核殼催化劑的Ru表面,并驗證了引入的活化羥基物種能直接促進堿性HER反應。獲得的R-Ru-Ni(OH)2催化劑展現出優異的堿性HER性能。原位光譜與質譜等分析證實了活化后的R-Ru-Ni(OH)2催化劑存在足夠多的羥基物種,并直接參與析氫反應;同時,作為優異的質子受體促進了水解離與*H脫附進程。DFT計算進一步驗證了羥基物種的形成改善了Ru活性位點的電子轉移能力,并優化了反應路徑與決速步驟;同時,Ru-OHad與界面水直接質子交換形成*H2O物種,裂解脫附后形成H2,從而極大促進了HER動力學。此外,該電化學激活策略亦表現出普適性。
文獻信息
https://doi.org/10.1021/acscatal.4c07602