原創 研理科研線-文獻 2025年09月20日 07:31北京
2025年9月15日,武漢理工大學木士春教授團隊在Nano Energy期刊發表題為“Suppressing high-valent ruthenium oxidation by oxide pathway mechanism for ultra-stable oxygen evolution”的研究論文,武漢理工大學焦吉祥、趙宏宇、陳釘為論文共同第一作者,華中科技大學涂正凱、武漢理工大學夏凡杰為論文共同通訊作者。
邏輯鏈條
綠色H2生產背景 → 堿性OER催化劑瓶頸(活性-穩定性矛盾)→ 核心問題(高價Ru活性高但不穩定,易溶解)→ 傳統反應路徑(AEM/LOM)的局限性 → 提出新思路(氧化物路徑機制OPM可兼顧活性與穩定性)→ 核心策略(Pr摻雜RuO2)→ 闡述機制鏈條(Pr提升Ru價態優化吸附 → 激活OPM路徑,避免晶格氧參與 → OH吸附后Ru價態降低,實現“自我穩定”,抑制過氧化和溶解)→ 實驗與理論證實超高活性與超長穩定性。
全文點評
讓我們來看看如何對這篇文獻深刻的拆解,清晰的呈現到你面前,制作不易,方便的話大家可以點贊轉發喲(公眾號加上星標,不迷路喲),尤其建議剛入門的研究生同學可以對著文獻和我們的解析進行對比閱讀,有利于打開大家的思路,理清邏輯思路。
話不多說,今天這篇文獻的主角,我相信做OER的老師同學都再熟悉不過了---RuO2。這個在催化界堪稱“勞模”的材料,關于它的活性-穩定性“蹺蹺板”難題,簡直是老生常談。文章發了無數,從調控結構到元素摻雜,各種“魔改”手段層出不窮。那么:作者是如何在這樣一個內卷到極致的體系上,還能講出新故事,并再次登上Nano Energy的呢?
這就引出了一個科研中非常重要的點:從“治標”到“治本”的思維躍遷。很多工作停留在‘頭痛醫頭腳痛醫腳’——Ru不穩定,我就加點別的元素把它‘按住’,但往往是以犧牲活性為代價。而這篇文章的聰明之處在于,它沒有滿足于‘按住’,而是思考:能不能讓Ru自己‘不想跑’?這就好比治水,堵不如疏。作者沒有單純去加固堤壩(增強結構穩定性),而是直接改了河道(切換反應路徑)!他們不再糾結于傳統的AEM或LOM機制的局限,而是將目光鎖定在一個更“先進”的反應路徑——氧化物路徑機制(OPM)上。這個機制本身就能避免晶格氧參與,從根源上降低了結構坍塌的風險。
本文的核心貢獻就在于,它不只是‘發現’了Pr摻雜能提高穩定性,而是清晰地‘設計’并‘證實’了Pr摻雜是為了激活OPM路徑。整個邏輯鏈條非常漂亮:Pr摻雜→提升Ru價態,優化初始吸附→引導反應走向OPM路徑→后續反應步驟中Ru價態反而降低,實現“自我保護”→最終達成活性和穩定性的雙贏。這套“組合拳”打下來,就把一個老大難問題給優雅地解決了。所以說,這篇文章的價值不在于又找到了一個性能不錯的RuO2基催化劑,而在于它提供了一種“機制引導設計”的新范式。它告訴我們,面對經典難題,與其在舊框架里修修補補,不如跳出來,從更底層的反應機理上尋找突破口。這才是從‘好工人’到‘巧工匠’的晉級之路。
理論計算分析
我們再看這篇文章的DFT理論計算部分,作者團隊的操作堪稱“釜底抽薪”式的機理剖析,精準地揭示了Pr摻雜為何能同時實現“高活性”與“超穩定”這對矛盾的統一。
首先,整個計算邏輯緊扣一個核心科學問題:Pr摻雜是如何在原子和電子層面,既保護高價Ru活性位點,又改變反應路徑,從而打破活性-穩定性“蹺蹺板”效應的?
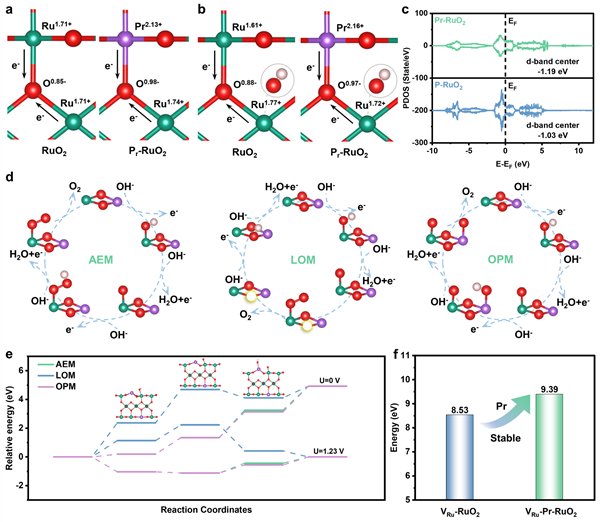
從源頭解釋“高活性”的由來(圖4a):通過Bader電荷分析,計算清晰地顯示,Pr原子取代Ru后,其周圍的Ru原子丟失了更多電子,呈現出更高的正電荷。這在理論上證實了Pr的引入提升了Ru的價態(與XAS實驗中+4.33的價態完美呼應),從而構建了具有優異本征活性的高價Ru位點。這是“高活性”的電子結構基礎。
揭示“超穩定”的核心奧秘——動態價態自調節(圖4b):這部分計算是全文理論部分的“點睛之筆”。作者對比了吸附關鍵中間體OH之后Ru位點的價態變化。對于純RuO2,Ru位點在吸附OH后價態進一步升高,離“過氧化溶解”更近一步。而對于Pr-RuO2,奇跡發生了:由于Pr-O-Ru微結構的存在,Pr和O原子會向Ru“反饋”電子,導致Ru位點的價態反而降低!這簡直是神來之筆,相當于給高活性的Ru位點上了一道“動態保險”,一旦開始反應,價態不升反降,從根本上避免了過氧化,解釋了“抑制高價釕氧化”的電子學起源。
優化中間體吸附,加速反應動力學(圖4c):通過計算態密度(PDOS),作者揭示了Pr-RuO2的d帶中心(-1.19 eV)相比RuO2(-1.03 eV)有所下移。根據Sabatier原理,過強的吸附不利于產物脫附。d帶中心的下移意味著對含氧中間體的吸附被適度削弱,這既有利于中間體的轉化,也防止了活性位點被“毒化”,從而提升了整體反應動力學。
鎖定最佳反應路徑,規避結構崩潰(圖4d, 4e):作者沒有滿足于單一路徑的計算,而是對比了AEM、LOM和OPM三種可能的OER路徑。計算結果明確指出,在Pr-RuO2表面,OPM(氧化物路徑機制)的速控步能壘最低(0.56 eV)。這意味著反應會優先走OPM路徑,即通過兩個吸附的O直接偶聯生成O2。這不僅本身能壘更低(高活性),更重要的是它避免了LOM路徑中晶格氧的參與,從而防止了催化劑的結構坍塌和降解(高穩定性)。
量化結構穩定性,提供直接證據(圖4f):為了進一步夯實“穩定性”的論點,作者計算了Ru原子的脫出能(demetallation energy)。結果顯示,Pr-RuO2表面Ru原子的脫出能比純RuO2高出的0.86 eV。這個巨大的能量差值強有力地證明了Pr原子像“鋼釘”一樣錨定了RuO?的晶格,極大地抑制了Ru的溶解和流失,為實驗中觀察到的1000小時超長穩定性提供了堅實的理論依據。
總而言之,這篇論文的DFT計算部分構建了一個從“電子調控”到“路徑選擇”再到“結構加固”的完美邏輯閉環。它深刻地闡明了Pr摻雜通過“靜態提價(高活性)+ 動態降價(防過氧)+ 路徑擇優(避坍塌)+ 晶格錨定(防流失)”這一整套組合拳,巧妙地解決了Ru基催化劑活性與穩定性的核心矛盾,是理論計算指導催化劑設計的絕佳范例。
摘要解析
摘要是文章的“精華濃縮版”,咱們按照“背景-問題-方案-亮點-意義”的框架來解析:
研究背景:在析氧反應(OER)中,高價態的Ru原子是公認的高效活性位點,它對反應中間體有極佳的吸附能。
存在的挑戰/問題:然而,這些高活性的高價Ru位點極不穩定,在電解水過程中容易被進一步氧化并溶解流失,導致催化劑失效,難以兼顧高活性和高穩定性。
本研究方案:作者提出了一種“氧化物路徑機制(OPM)”策略來保護高價Ru位點。具體方法是采用熔鹽法,將鐠(Pr)原子摻入RuO2納米顆粒中(形成Pr-RuO2)。
核心亮點與結果:
1.高活性來源:Pr的引入成功將Ru的價態提升至+4.33,優化了對反應中間體的結合能。
2.高穩定性機理:反應開始后(形成OH),Ru位點的價態反而會降低,從而穩定了活性位點。更重要的是,原位光譜/質譜和理論分析證實,Pr-RuO2激活了OPM反應路徑,同時提高了Ru空位的形成能,避免了結構坍塌和催化劑失效。
3.優異性能:Pr-RuO2在10 mA cm-2電流密度下僅需216 mV的過電位,其質量活性是商業RuO2的13.4倍。在三電極體系中穩定運行長達1000小時,在陰離子交換膜水電解槽(AEMWEs)中也表現優異。
研究意義:這項工作通過調控反應路徑,成功地保護了高活性的高價Ru位點,為解決RuO?基催化劑的穩定性難題提供了全新的思路,并有望推動AEMWEs技術在大規模制氫中的應用。
研究亮點與數據支撐(證據鏈)
本研究的創新解決之處在于,它沒有像傳統方法那樣通過降低Ru價態來犧牲活性換取穩定性,而是反其道而行之:先構建更高價、更高活性的Ru位點,然后通過巧妙的“機制工程”(誘導OPM路徑)來為這個高活性位點提供“動態保護”,從而一舉打破了活性-穩定性的“蹺蹺板”限制。
證據一(高價Ru活性位點的成功構筑):
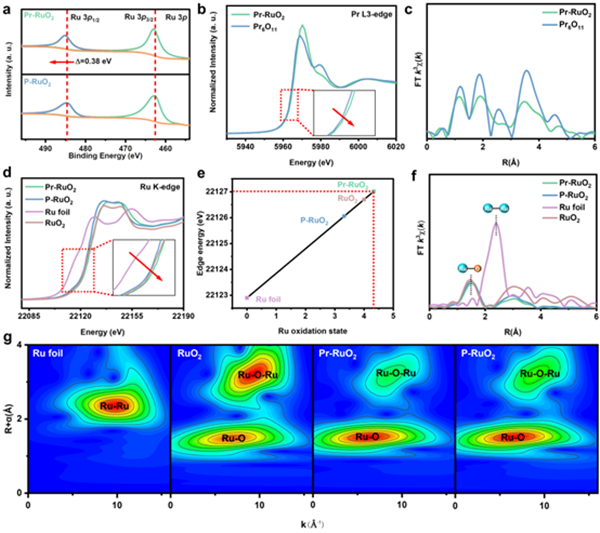
XPS & XAS光譜(圖2):這是證明“高價Ru”存在的直接證據。XPS光譜(圖2a)顯示Pr-RuO2的Ru 3p峰向高結合能方向移動,表明Ru的氧化態升高。更定量的Ru K邊XAS譜(圖2e)通過線性擬合,精確計算出Pr-RuO2中Ru的平均價態高達+4.33,遠高于純RuO2,證實了高活性位點的成功構建。
證據二(OPM反應路徑的“實錘”證據):
18O同位素標記DEMS實驗(圖5c, 5d): 這是區分反應路徑的“金標準”。實驗中,Pr-RuO2在反應中檢測到了明顯的18O18O信號,這直接來源于兩個吸附的18O原子之間的直接偶聯,是OPM路徑的決定性證據。相比之下,P-RuO2則沒有此信號,表明其遵循不同機制。
原位ATR-FTIR光譜(圖5f):該光譜在Pr-RuO2表面檢測到了歸屬于-O-O-(過氧/超氧物種)的特征峰,這是OPM路徑中關鍵中間體O2的存在證據,進一步佐證了OPM機制。
證據三(反應過程中活性位點的“動態保護”):
DFT計算(圖4b):理論上揭示了保護機制。計算表明,當OH吸附在Pr-RuO2的Ru位點上時,Ru的價態會不升反降,從而避免了向更高價態(如RuO2)的過氧化,這是穩定性的根本原因。
原位拉曼光譜(圖5g, 5h):實驗上觀測到了結構的穩定性。在工作電位下,Pr-RuO2的拉曼峰位基本不變,表明其結構穩定。而對照組P-RuO2在1.4V以上出現了代表更高價Ru物種的新峰(780 cm-1),表明其發生了不穩定的表面氧化。
證據四(策略成功的宏觀體現):
超長穩定性測試(圖3d):Pr-RuO2在10 mA cm-2下穩定運行了1000小時,衰減率極低(23.3 μV s-1),這是對其超高穩定性的最直觀展示。
ICP-OES分析:經過20小時OER測試后,Pr-RuO2中Ru的溶解量僅為0.2%,遠低于P-RuO2的1.6%,從原子層面證實了其優異的抗流失能力。
前言解析
-
前言部分,作者為我們清晰地勾勒出其研究的邏輯脈絡:
開篇點題,指出氫能作為未來清潔能源戰略的關鍵,而通過可再生能源驅動電解水是實現“綠氫”生產的核心路徑。接著,作者將焦點縮小至堿性條件下的陰離子交換膜水電解槽(AEMWEs),肯定其商業化潛力,但同時直指其陽極催化劑活性不足和穩定性差的“痛點”。 由此,作者自然地引出了主角——二氧化釕(RuO2),并闡明其作為AEMWEs陽極催化劑的優勢。緊接著,文章切入核心矛盾:高價態的Ru物種雖然具有更高的本征催化活性,但在氧化電位下極易被過度氧化而溶解,導致催化劑失效,這便是著名的“活性-穩定性”蹺蹺板難題。作者簡要回顧了前人試圖通過降低Ru價態來提升穩定性的策略,并指出這種以犧牲活性為代價的方案并非最優解。 話鋒一轉,作者提出了破局之道——通過調控反應路徑來打破“活性-穩定性”的制衡。
-
在對比了傳統的吸附演化機制(AEM)和晶格氧機制(LOM)的利弊后,作者亮出了他們的核心創意:引入最近報道的“氧化物路徑機制”(Oxide Pathway Mechanism, OPM)。該機制通過O中間體的直接耦合生成O2,既避免了形成高能壘的OOH中間體,也繞開了導致結構坍塌的晶格氧參與,有望同時實現高活性與高穩定性。
-
最后,作者明確闡述了本研究的核心論點:通過將鐠(Pr)原子摻入RuO2納米晶中,可以激活OPM路徑,從而保護高活性的高價態Ru位點。具體來說,Pr的引入不僅將Ru的價態提升至+4.33,優化了中間體的吸附能(提升活性);更關鍵的是,它改變了反應路徑,在OH吸附后反而降低了Ru的價態,并通過激活OPM路徑抑制了晶格氧的參與,從根本上防止了Ru的溶解和結構崩潰(提升穩定性)。
圖文對照解析
圖1 材料的合成與表征:證實了通過熔融鹽法成功制備了Pr原子摻雜的、具有特定微觀結構的Pr-RuO2納米催化劑。
-
a 示意圖展示了Pr-RuO2的熔融鹽合成路線及其催化過程。
-
b, c TEM和高分辨TEM圖像顯示,與商業和純RuO2相比,Pr-RuO2的顆粒尺寸更小(平均3.81 nm),且結晶性良好。
-
d, e EDS和EELS元素分布圖譜證實Pr、Ru、O元素在納米顆粒中均勻分布。
-
f, g 球差校正HAADF-STEM圖像及其原子強度分析直觀地證明了Pr原子成功取代了RuO2晶格中的部分Ru位點。
-
h XRD圖譜表明,Pr的摻雜沒有改變RuO2的金紅石晶體結構。
圖2 催化劑的電子結構分析:揭示了Pr的引入如何調控了Ru的電子結構,使其處于高氧化態。
-
a Ru 3p的XPS譜圖顯示,與P-RuO2相比,Pr-RuO2的峰位向高結合能方向移動,表明其Ru的氧化態更高。
-
b, c Pr L3邊的XAS譜圖分析表明,Pr-RuO2中的Pr主要以Pr3+和Pr4+混合價態存在,且其配位環境與Pr6O11有顯著差異。
-
d, e Ru K邊的XANES譜圖及線性擬合結果定量地指出,Pr-RuO2中Ru的平均價態高達+4.33,顯著高于P-RuO2(+3.32)和標準RuO2。
-
f, g Ru K邊的EXAFS譜圖及其小波變換分析顯示,Pr-RuO2的近鄰配位結構與標準RuO2幾乎一致,再次證實了Pr的晶格取代。
圖3 OER催化性能與AEMWEs器件性能:全面展示了Pr-RuO2優異的催化活性、動力學和穩定性。
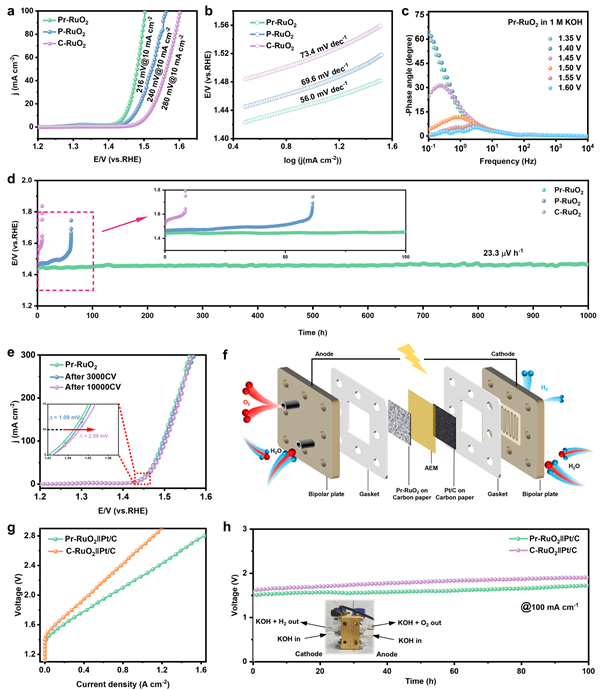 a, b LSV曲線和Tafel斜率圖顯示,Pr-RuO2在10 mA cm-2下僅需216 mV的過電位,且具有更快的反應動力學(Tafel斜率56.0 mV dec-1)。 c Operando EIS的Bode圖表明,在工作電位下,Pr-RuO2具有更快的電荷轉移速率。
a, b LSV曲線和Tafel斜率圖顯示,Pr-RuO2在10 mA cm-2下僅需216 mV的過電位,且具有更快的反應動力學(Tafel斜率56.0 mV dec-1)。 c Operando EIS的Bode圖表明,在工作電位下,Pr-RuO2具有更快的電荷轉移速率。
-
d 計時電位測試結果驚人地顯示,Pr-RuO2在10 mA cm-2的電流密度下穩定運行超過1000小時,衰減速率極低。
-
e 加速穩定性測試(10000次CV循環后)進一步證實了其卓越的結構穩定性。
-
f, g, h 組裝的AEMWEs器件測試表明,Pr-RuO2作為陽極,在60 °C、100 mA cm-2電流密度下僅需1.52 V電壓,并能穩定運行100小時,展現了巨大的實際應用潛力。
圖4 DFT理論計算:從理論層面揭示了Pr摻雜提升活性與穩定性的內在機理。
-
a, b Bader電荷分析顯示,Pr的引入提升了初始Ru位點的價態,但在吸附OH中間體后,反而通過Pr-O-Ru微結構使Ru價態降低,從而避免了過度氧化。
-
c 態密度(PDOS)計算表明,Pr-RuO2的Ru d帶中心更低,有利于削弱中間體吸附。 d, e OER路徑的自由能計算表明,對于Pr-RuO2,OPM是能量上最有利的反應路徑,其速控步能壘(0.56 eV)低于AEM和LOM。
-
f 脫金屬能計算顯示,Pr-RuO2的Ru原子脫出能壘遠高于RuO2,表明Pr的引入顯著增強了結構的穩定性。
圖5 催化機理分析:通過一系列原位表征和實驗證據,證實了Pr-RuO2遵循OPM反應路徑。
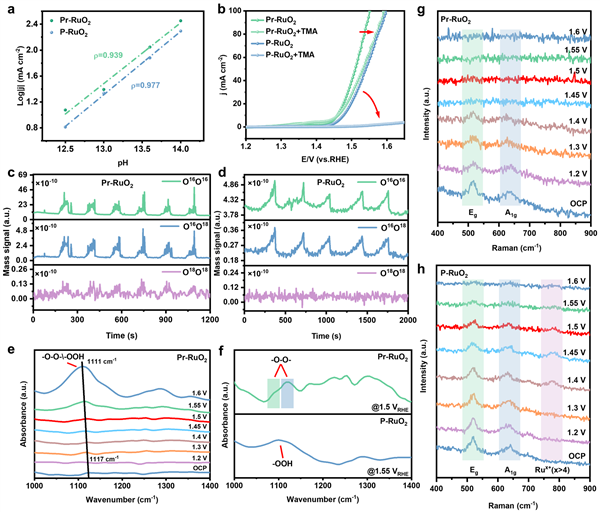
a, b pH依賴性實驗和TMA+毒化實驗初步表明,Pr-RuO2的反應路徑不同于以LOM為主的P-RuO2。
-
c, d 18O同位素標記的DEMS實驗是關鍵證據:Pr-RuO2檢測到了18O18O信號,這是兩個吸附的18O原子直接耦合(OPM路徑的標志)產生的,而P-RuO2則沒有此信號。
-
e, f 原位ATR-SEIRAS光譜在Pr-RuO2表面檢測到了歸屬于-O-O-中間體的振動峰,為OPM路徑提供了進一步支持。
-
g, h 原位拉曼光譜顯示,在OER過程中,Pr-RuO2的結構比P-RuO2穩定得多,后者在較高電位下出現了與高價Ru物種相關的峰,預示著其不穩定性。
最后點評
這篇文章是一項極為出色和深刻的研究工作。它巧妙地利用“鐠(Pr)摻雜”這一策略,精準地解決了長期困擾RuO2基催化劑的“活性-穩定性”蹺蹺板難題。該工作的核心貢獻在于,它不僅合成了一種性能卓越的催化劑,更重要的是,通過DFT理論計算結合多種精巧的原位譜學/質譜聯用技術,令人信服地揭示了其背后的深層機制:Pr的引入通過調控電子結構,將OER反應路徑從易導致結構崩潰的傳統機制引導至更為穩定且高效的“氧化物路徑機制”(OPM),從而在高價Ru位點提供高活性的同時,又避免了其在反應中被過度氧化和溶解。這項發現不僅為設計超穩定貴金屬OER催化劑提供了全新的、可行的思路,也加深了我們對催化反應路徑調控重要性的理解,為在原子和電子層面理性設計高性能能源材料開辟了新的視角。
文獻引用:
[1] J. Jiao, H. Zhao, D. Chen, X. Cao, X. Luo, H. Wu, L. Jiang, J. Zhang, B. Tian, Z. Tu, W. Zeng, F. Xia, S. Mu, Suppressing high-valent ruthenium oxidation by oxide pathway mechanism for ultra-stable oxygen evolution. Nano Energy 2025, 145, 111468.
文章鏈接:https://doi.org/10.1016/j.nanoen.2025.111468
作者信息
木士春:武漢理工大學首席教授,博士生導師,國家級高層次人才。長期致力于質子交換膜燃電池和電解水催化劑研究。以第一作者或通訊作者在Nat. Commun.、Adv. Mater.、J. Am. Chem. Soc.、Angew. Chem. Int. Ed.、Energy Environ. Sci.、Nano Lett.等國內外期刊上發表300余篇高質量學術論文。
Email:[email protected]
[團隊介紹]
武漢理工大學先進能源材料研究團隊依托材料復合新技術國家重點實驗室,長期從事質子交換膜燃料電池關鍵材料與核心器件、電化學產氫催化材料、鋰離子電池電極材料和碳納米材料等研究工作。歡迎有志于科技報國的研究生及博士后加入團隊!
木士春研究團隊主頁http://shichunmu.polymer.cn/