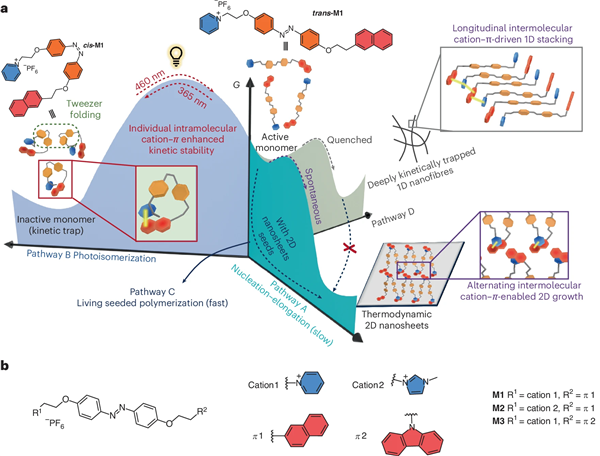
超分子聚合作為一種廣泛使用的合成方法,在構建復雜納米結構方面展現出巨大潛力,然而在聚合過程與結構的精確控制上仍明顯落后于生物分子系統,根源在于多數超分子聚合過程具有自發性,導致對聚合進程與聚合物結構的精準調控存在困難。近年來,活性超分子聚合作為可精確控制聚合物長度、多分散性與序列結構的策略備受關注,但如何有效耦合動力學陷阱與熱力學聚合以構建一體化能量景觀仍具挑戰。傳統非共價相互作用(如氫鍵)雖常用于路徑調控,但其對環境高度敏感且方向性單一,使動力學勢壘高度依賴亞穩態鍵合模式的環境穩定性,且易出現陷阱過深或過淺而難以與后續熱力學路徑有效銜接,同時也限制了高維有序熱力學產物的形成。因此,通過設計兼具特定非共價識別基元與潛在構象轉換能力的單體,并在動力學陷阱與熱力學路徑的耦合間實現微妙平衡,是解決上述問題的有效途徑。
近日,課題組提出了以芳香陽離子–π相互作用為主導的路徑調控新策略:通過動態切換分子內/分子間陽離子–π鍵合模式并結合偶氮苯光異構化,實現動力學陷阱與熱力學聚合之間的能量景觀耦合。首先設計并合成了以反式偶氮苯為核心、兩端分別修飾芳香陽離子與π單元的線性單體M1–M3;其次反式-M1可通過交替的分子間陽離子–π相互作用自發“成核–延伸”聚合,形成有序二維納米片(路徑A);再次在365 nm光照下,單體折疊并由分子內陽離子–π作用穩定為非活性構象,進入動力學陷阱(路徑B);最后當切換至460 nm光照后,構象展開釋放可聚合活性單體,并在二維種子存在下實現快速、種子引發的活性超分子聚合(路徑C)。此外,快速淬火也可獲得處于深動力學陷阱的一維納米纖維(路徑D)。該策略可進一步實現對電催化析氫反應的原位調控,聚合過程中過電位由210 mV降至123 mV,Tafel斜率降至75.8 mV/dec,展示了陽離子-π作用主導的活性超分子聚合過程在對功能材料性能原位調控中的潛力。
相關研究成果以“Aromatic cation-π-dominated pathway control in living supramolecular polymerization”為題發表于《Nature Synthesis》(Nat. Synth. 2025, DOI:10.1038/s44160-025-00912-6)。課題組博士研究生張哲霖和蘇俊龍碩士為論文的共同第一作者,田威教授為論文通訊作者。
(全文鏈接:https://doi.org/10.1038/s44160-025-00912-6)。