Finally, someone has explained the principle of adsorbate evolution mechanism (AEM)!
原創 GBZ電化學與電催化2025年07月15日15:04廣東
說明:本文系統闡述了析氧反應(OER)中的吸附質演化機制(AEM),包括其原理、研究意義及應用進展研究。


什么是吸附質演化機制(AEM)
析氧反應(OER)作為關鍵電極反應,在電解水制氫、金屬空氣電池和可再生燃料合成等能源轉換體系中發揮核心作用。OER涉及復雜的多電子轉移和含氧中間體的轉換,導致其動力學過程緩慢,通常伴隨著較高的過電位。
隨著催化科學的發展, OER機制研究已從早期的吸附演化機制(AEM)和晶格氧機制(LOM)擴展至多個新興理論,如氧化物路徑機制(OPM)、氧-氧耦合機制(OCM)和分子內氧耦合機制(IMOC)。
然而,OER機制的復雜性不僅體現在不同催化劑體系間的機制差異,還涉及催化過程中不同機制的協同作用和動態轉變。厘清OER機制的本質、建立精確的機制調控策略,是開發高效、穩定OER催化劑的核心科學問題,對推進電催化能源技術的工業應用具有重要意義。


AEM機制的原理
提高OER過程中的電化學動力學可通過加速催化劑和氧氣中間體之間快速的電子轉移實現,這很大程度上與費米能級周圍的電子態有關。費米能級附近電子態表現為金屬特性時,表現為金屬為氧化還原中的AEM機制;費米能級附近電子態表現為氧時,表現為氧為氧化還原中的LOM機制。
吸附質進化機制(AEM)包括氧中間體經歷吸附、去質子化、偶聯和解吸(*OH→*O→*OOH→O2)的順序過程。同樣,在不同的電解液環境下OER也會發生不同的反應,如圖1。
酸性環境下:
*+H2O→HO*+H++ e-
HO*→O*+H++ e-
O*→1/2O2+*
H2O+O*→HOO*+H++ e-
HOO*→O2+H++ e-
堿性環境下:
*+OH-→OH*+ e-
HO*+OH-→O*+H2O+ e-
O*→1/2O2+*
O*+OH-→HOO*+ e-
HOO*+OH-→O2+H2O+ e-
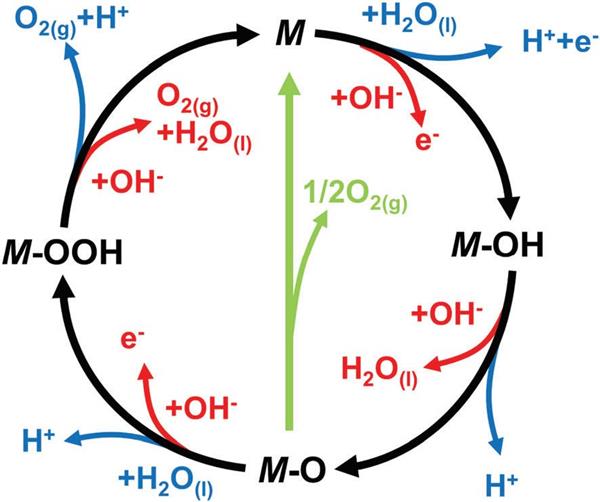
圖1. OER在酸性(藍色箭頭)、堿性環境(紅色箭頭)下的AEM機制(黑色箭頭代表OER過程中可能涉及的中間體的形成,綠色箭頭表示O2生成的另一種途徑)。DOI:10.1039/C6CS00328A


AEM機制的研究意義
根據Sabatier原理,性能優異的催化劑取決于催化位點和氧中間體(*OH、*O、*OOH)結合能之間的平衡。在異相催化/電化學中的標度關系(Scaling Relation)指出不同物理量在一系列催化體系上具有線性相關關系。
在OER中,ΔGHO*、ΔGHOO*分別與ΔGO*存在標度關系,所以ΔGHO*與ΔGHOO*之間也存在標度關系,并推斷出ΔG(*OOH)-ΔG(*OH)= 3.2 ± 0.2 eV。
如果在某種催化劑上ΔGHO*→O*降低,那么ΔGO*→HOO*升高,反之亦然。當ΔGHO*→O*和ΔGO*→HOO*相等時,催化劑的吸附位點最接近火山圖峰值時,催化劑的效率最高(如圖2),產生最小的過電位,可以得出AEM途徑的理論極限過電位為370 mV。AEM途徑作為OER中具有基礎性、普適性和理論奠基作用,是理解OER過程的核心框架。
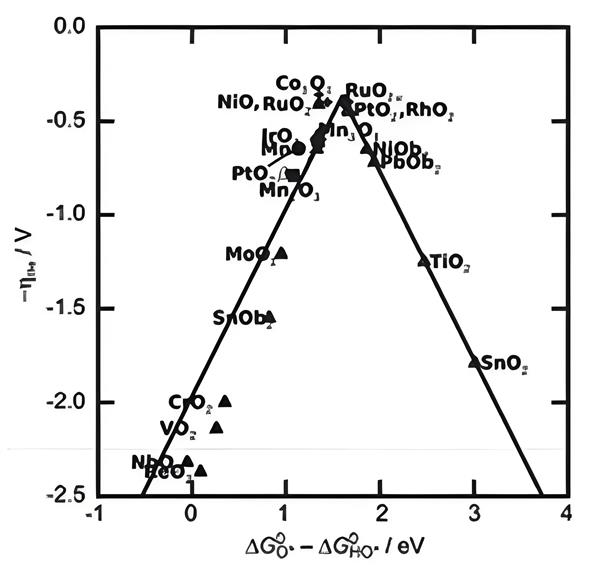
圖2. 不同金屬氧化物的OER活性趨勢(使用吸附能作為描述符)。DOI:10.1002/smll.202202336


AEM機制的應用
案例一:在不改變AEM機制的前提下,通過催化劑的改性策略,增強反應動力學突破其理論極限過電位(370 mV)。
2025年4月17日,蘭州大學李澤龍、香港城市大學黃勃龍、中國科學院上海應用物理研究所李炯在國際知名期刊Advanced Materials發表題為《Fluorine Engineering Induces Phase Transformation in NiCo2O4 for Enhanced Active Motifs Formation in Oxygen Evolution Reaction》的研究論文。
作者報道了氟(F)摻雜的NiCo2O4(NiCo2O4-Fn)OER催化劑,該催化劑由NiCo2O4核和(NH4)NixCo1-xF3外殼組成,NiCo2O4-F1的本征活性比NiCo2O4提高了14倍。
密度泛函理論(DFT)計算表明,F摻雜導致了大量氧空位的產生(Ov),Ov誘導的不飽和Co和Ni位點通過促進*OH中間體吸附和轉化,降低了OER的能壘,從而增強了電活性,其電化學性能如圖3所示。
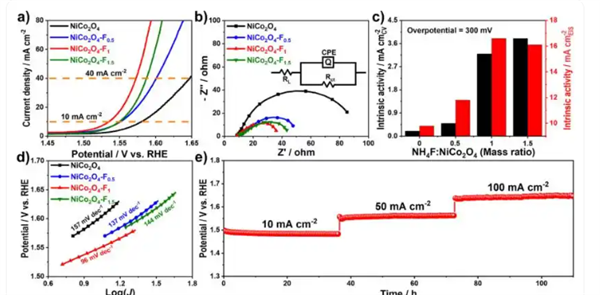
圖3. 催化劑的電化學性能測試。DOI:10.1002/adma.202418058
如圖4所示,同位素標記的差分電化學質譜(DEMS)測量結果表明,18O標記NiCo2O4-F1和18O標記NiCo2O4的32O2 - 34O2 - 36O2記錄的質量信號比值分別為1804:27:1和3645:27:0。這表明NiCo2O4和NiCo2O4- F1都遵循吸附質演化機制(AEM)催化OER。即該催化劑設計策略在不改變AEM反應機制的同時增加了電化學活性。
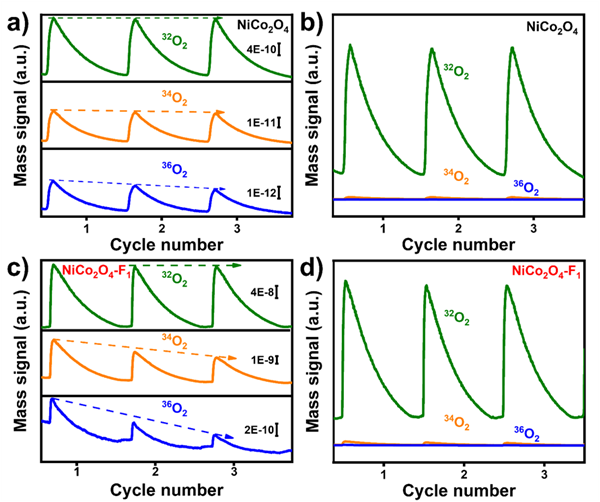
圖4. 同位素標記的原位差分電化學質譜圖像DOI:10.1002/adma.202418058
案例二:通過構建催化劑雙活性位點,將AEM途徑和有更高動力學的途徑(如LOM等途徑)相結合,實現雙機制共存策略。
2024年9月18日,武漢理工大學木士春團隊在Nature Communications上發表題為《Fe-S dually modulated adsorbate evolution and lattice oxygen compatible mechanism for water oxidation》的研究論文。
作者以NiMoO4水合物為預催化劑,Fe和S為調節劑,通過化學腐蝕共同引入,誘導了大量的結構缺陷,促進了電化學活化過程中完全重構為R-NiFeOOH@SO4。陽極活化觸發金屬和晶格氧中心的氧化還原,并涉及氧空位的形成和重新填充。
如圖5所示,R-NiFeOOH@SO4催化劑在1.0 M KOH溶液中達到100 mA cm-2電流密度所需的過電位為251±5 mV。

圖5. 催化劑的電化學性能測試。DOI:10.1038/s41467-024-52682-y
在圖6a中,所有催化劑的OER活性都表現出一定程度的pH依賴性,而R-NiFeOOH@SO4的電流密度隨著pH值的降低而下降得更快,這表明晶格氧參與了OER過程。
此外,在圖6b中,R-NiFeOOH@SO4在1M TMAOH中,由于四烷基銨陽離子(TMA+)的強結合抑制了LOM, OER活性明顯減弱。而對R-NiFeOOH和R-NiOOH只有輕微的活性衰減。
為了進一步驗證催化劑晶格氧在OER過程中的參與,采用了原位18O同位素標記差分電化學質譜法(DEMS)。結果表明其具有更高的晶格氧活性和更有利的LOM通路。同時,與RNiOOH相比,R-NiFeOOH的16O18O峰面積增大,而其16O18 O/ 16O16 O峰面積比值減小,說明在AEM途徑下,Fe的電子調控對析氧的貢獻更為顯著。此外,用18O同位素標記的原位拉曼光譜也證實了晶格氧參與了OER過程。
如圖6d-f所示,在氧質量對振動模式的影響下,18O標記的催化劑明顯向低波數偏移。在含16O的電解液中,以1.624 V / RHE恒定電位作用時,隨著18O的消耗,18 O標記催化劑的拉曼峰逐漸移回常規16O標記催化劑的位置,其中R-NiFeOOH@SO4晶格氧釋放更快,拉曼峰在1 min內移回。R-NiFeOOH和R-NiOOH需要20min甚至更長的時間來釋放標記的18O。這一結果進一步證實了R-NiFeOOH@SO4在OER過程中更快的晶格氧釋放,揭示了S調制對晶格氧的有效活化。
在圖6g-i中,隨著施加電位的增加,可以在1200和1100 cm?1附近觀察到明顯的吸收峰,表明產生了含氧中間體。位于1207-1212 cm-1的峰可歸因于在LOM途徑中形成的O-O中間體,進一步證實了LOM的參與。
同時,在1095 cm?1處的峰屬于AEM途徑產生的*OOH。與R-NiOOH相比,R-NiFeOOH的*OOH峰顯著增強,O - O峰明顯減弱,表明Fe引入對AEM途徑OER進行了優化。
在圖6i中,R-NiFeOOH@SO4在1115 cm?1處呈現合并且強度更高的* OOH峰,并且O-O峰進一步增強,表明AEM和LOM同時優化。原位ATR FTIR結果進一步證實了催化劑AEM和LOM的耦合作用,其中R-NiFeOOH@SO4通過協同優化AEM和LOM促進了OER過程。
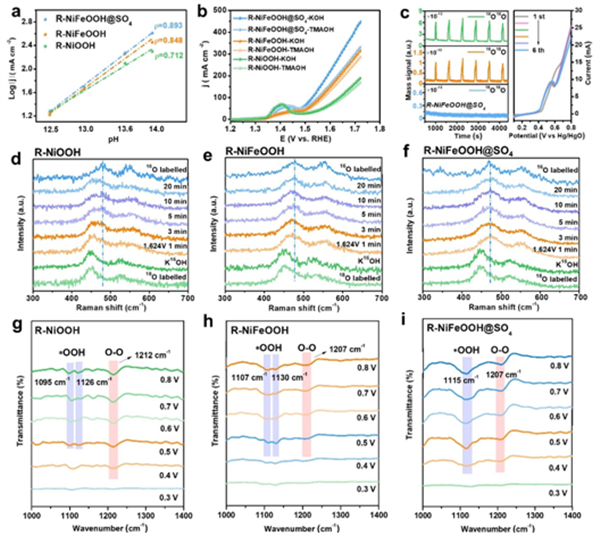
圖6. AEM-LOM兼容機制OER催化分析。包括電流的pH依賴性測試、同位素標記的原位差分電化學質譜分析、拉曼分析和紅外分析。DOI:10.1038/s41467-024-52682-y
案例三:以AEM途徑作為OER反應的基礎途徑,與文章中新提出的反應途徑作為對比(如OPM、OCM、IMOC等途徑),突出新機制途徑的優勢。
2024年2月29日,山東大學王建軍教授、宋克鵬教授在JACS上發表題為《Ir Single Atoms Boost Metal?Oxygen Covalency on SelenideDerived NiOOH for Direct Intramolecular Oxygen Coupling》的研究論文。
作者將Ir單原子錨定在NiOOH上(Ir1@NiOOH),其中Ir取代前驅體FeNiSe2晶格中的Fe,促進硒化物向活性結構羥基氧化物的轉變。這種元素取代促進了O 2p帶的上移,增強了金屬-氧共價,從而實現了OER機制的轉變,使其活性增強。
原位差分電化學質譜(DEMS)證實了NiOOH的單金屬位點機理到Ir1@NiOOH的雙金屬位點機理的轉變,其間涉及的原子級結構和螯合配位環境的轉變獲得了原位拉曼、X射線吸收光譜和理論計算結果的支持。
Ir1@NiOOH電極表現出優異的電催化性能,在電流密度為10和1000 mA cm-2時分別實現了低至142和308 mV的過電位,為OER的電極設計設定了新的基準。
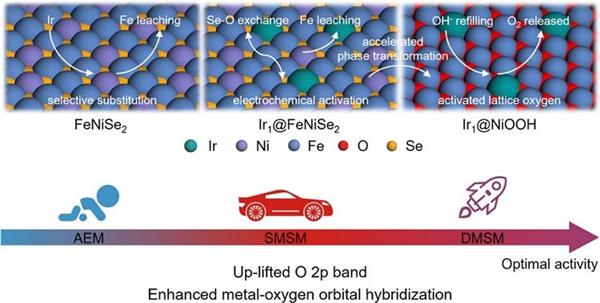
圖7. Ir1@NiOOH通過選擇性取代和電化學活化原理圖。DOI:10.1021/jacs.3c13746


總結
AEM是理解OER的基礎,但通過缺陷工程、元素摻雜或機制協同可優化其動力學性能,甚至衍生新機制以突破理論極限。